6.9 Add hydrogens
In order to add hydrogens to a
molecule or to change the ionization state, you can use the following dialog box that can be shown by Edit
![]() Add
Add
![]() Hydrogens menu item:
Hydrogens menu item:
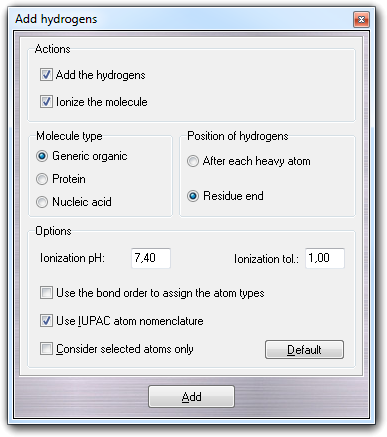
6.9.1 Dialog options
In Actions box, you can choose to add the hydrogens and/or to ionize the molecule in the current workspace. You must remember that if you want to ionize, the molecule must have the hydrogens and the algorithm add/remove the hydrogens according to the predicted pKa values and the specified pH.
In Molecule type box, you can select the molecule type (Generic organic,
Protein, Nucleic acid), in Position of hydrogens, you can
select where the hydrogens will be placed in the molecule file (After each heavy atom or at Residue end)
and in Options, you can also choose the nomenclature type used for the
hydrogen atoms (normal progressive
number or IUPAC nomenclature), the possibility to process the selected atoms only (Consider
selected atoms only) and algorithm used to add the hydrogens. More in
detail, if you check Use the bond
order to assign the atom types, instead of the bond geometry, the bond order
is not used to detect the atom hybridization. This feature is useful when the molecule has uncertain geometry (e.g.
2D instead of 3D structure) and the bond order is correctly assigned. When you
enable the ionization feature by checking Ionize the molecule, you can
also specify the pH of the solution (Ionization pH) in which the molecule
is virtually present as solute and the tolerance (Ionization tol., in pH
units) used to establish which protonation state is largely present. To this
end, the Henderson-Hasselbalch equation is used: if you specify 1 as ionization
tolerance, it means that, at the specified pH value, the concentration of the
ionized form must be at least 10 times higher than the non-ionized one to
consider the molecule as ionized.
Finally, you must click Add to
add the hydrogens and/or to ionize the molecule.
6.9.2 About the method to add the hydrogens
To add the hydrogens, the recognition of atom types and valences is
needed. The powerful ATDL geometry-independent engine can't be used because the atom valences are
incomplete due to the missing hydrogens and so the implemented method is based
on the original code included in Babel 1.6 (© 1992-96, W. Patrick Walters, Matthew T.
Stahl) that detects the atom valences considering the atomic distances and the
bond angles. Unluckily, this is the method limit, because some structures don't
have a canonical geometry and so the valence detection can be wrong. VEGA
introduces an algorithm that fixes the atom type and the valence detection when
the user specify the type of the molecule. You must remember that this check
is disabled selecting Generic organic molecule type.
The method can be summarized in the following steps:
All hydrogens are removed in order to allow the correct atom type detection.
Atom type and valence attribution: the atom types are detected using the Meng convention (E. Meng and R. Lewis, J. Comp. Chem., 12, pp 891-898, 1991) updating it with Nim atom type to detect the nitrogens in five member rings.
Optionally, to fix the incorrect atom types, VEGA ZZ uses two templates (NA.hyb for nucleic acids and PROTEIN.hyb for proteins) placed in the Data directory. They are text files witch format is explained in the header.
The required hydrogens are placed and bonded.
6.9.3 About the method to ionize the molecule
To predict the ionization state of a molecule, the knowledge of the pKa values of each moiety included in its structure is needed. At this time, it was implemented a fast method based on the recognition of acid and basic groups whose pKa values are known. The Handerson-Hasselbach equation is used to evaluate the ratio between the acid/base pairs at the specified pH value and the group is considered ionized only if the this ratio is grater than the ionization tolerance defined by the user. The algorithms is so summarized:
All these steps are repeated until there are no hydrogens to add/remove.