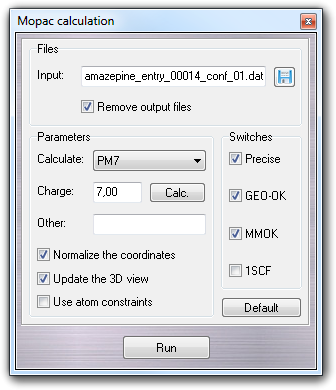
8.4 Mopac calculation
Through this dialog window, you can run Mopac
semi-empirical calculations. Press Run button to start the calculation
using the Mopac
executables included in the standard VEGA ZZ package. If you want to use another Mopac version, you must
replace the included executables placed in the VEGA ZZ installation directory
(usually ...\VEGA ZZ\Bin\Win32). The
Input field is automatically filled by VEGA ZZ with the full file name of
the input file that will be created to run Mopac. In the same directory, Mopac
will generate the output files that will be automatically removed if Remove
output files is checked. If you want to place the output in another
directory, you can change the Input field by clicking the disk button or
by typing manually the file name with full path.
Mopac runs in background and VEGA ZZ remains fully functional. When the calculation
ends without errors, the optimized structure is automatically loaded. To abort a calculation,
you can use the Task Manager or select Stop calculation
in the context menu shown by clicking on the workspace.
Checking Update the 3D view, available only if Mopac2007/2009/2012 is installed, the
molecule structure is automatically updated during the calculation and the
progress is shown in the console (step number, energy and gradient).
Checking Normalize the coordinates, the molecule is translated at the
origin of Cartesian axis when the calculation is finished. Finally, Default
button allows to revert to pre-defined settings.
Checking Use atom constraints, available only if Mopac2007/2009/2012 is installed, you can fix some atoms that aren't moved during the optimization. To apply the atom constraints, you can use the Constraint options dialog box.
8.4.1 Parameter fields
By these fields, you can specify the type of
calculation (AM1, MINDO/3, MNDO, PM3; MNDOD, PM6, RM1 if Mopac2007 is installed, MOZYME, MOZYME RAPID, if Mopac2009 is installed
and PM7, PM7-TS, if Mopac2012 is installed), the total charge (if the pre-calculated charge is
wrong) and the extra keywords (Other field). The default parameter is
AM1 and the total charge that is calculated using the atomic partial charges if
they are already assigned, otherwise is calculated by recognition of charge groups.
It can be possible that the molecule could have wrong or incomplete atomic
charges and so a wrong value is shown. To force the group-based total charge
calculation, you can click Calc. button.
When Mopac2007/2009/2012 is installed, it's automatically recognized and the
graphic interface adapts itself to the new features (e.g. PM6, RM1 etc). Mopac2007
or greater isn't included in the VEGA ZZ package and it can be installed following
these steps.
8.4.2 Switches
They are the main switches for Mopac calculation. To understand what they mean, please read the Mopac Manual.
| Switch | Description |
| Precise | Increase the criteria for terminating all optimizations by a factor, normally 100. |
| GEO-OK | Override some safety checks (e.g. in the structure geometry). |
| MMOK | Use the molecular mechanics correction for the amidic bonds (CONH). It can be automatically switched on if an amidic bond is detected. |
| 1SCF | Do a single point energy calculation. |
8.4.3 Limits
VEGA ZZ includes two versions of Mopac 7.01-4: Mopac_50_100.exe (max 50 heavy atoms and max 100 hydrogens) and Mopac_100_200.exe (max 100 heavy atoms and max 200 hydrogens). The two executables are selected automatically by VEGA ZZ for a balanced use of the hardware resources. No more than 300 atoms (100 heavy atoms + 200 hydrogens) can be used by VEGA ZZ in Mopac calculations. These limits can be superseded installing Mopac2007 or greater (for more information, click here).