12.8 MetaQSAR - Management system for metabolic reactions
Index
12.8.2.4.1 Search by similarity
Drug metabolism with its many enzymes and reactions is a key factor in early ADMET screens for the selection of promising drug candidates, but its success depends on the reliability of available tools. With a view to supporting metabolic screening, MetaQSAR was developed in order to collect and classify metabolic reactions. The metabolic data can be retrieved from in-home experiments and/or meta-analysis of the literature and can be used for different kind of studies such as: analysis of the property space of the metabolic substrates, prediction of the metabolism and prediction of the production of toxic metabolites.
MetaQSAR plug-in offers a complete input system to manage and classify not only the metabolic reactions, but also the related substrate and products in term of molecular properties and 1D, 2D and 3D structures. To do that, MetaQSAR uses the multi-purpose VEGA ZZ database engine that offers different database engines (such as Access, MySQL, SQLite, SQL Server and more in general all ODBC data sources) able to manage in easy way a large number of molecules.
MetaQSAR plug-in is a complete system for the management of metabolic reactions. To use
it, you must show the main window, selecting Tools
![]() MetaQSAR in VEGA ZZ main menu.
MetaQSAR in VEGA ZZ main menu.
At the top of this window, there is the menu bar whose functions are summarized in the following table:
|
Item levels |
Shortcut | Description | ||||||||||||||||||||||
| 1 | 2 | 3 | ||||||||||||||||||||||
| File | New database | - | Ctrl+N | Create a new empty MetaQSAR database.
You can choose the format between Access 2003 (mdb) and Access 2007 (accdb).
MetaQSAR supports also MySQL data sources, but in this case the best way
is the use of a tool for the database conversion such as
Access to MySQL tool. To connect to the database, you can use this
data source file, that must be saved with .dsn file extension (e.g.
MySQL MetaQSAR.dsn):
MySQL ODBC connectors for Windows are available here. Download and install x86 32 bit version (MSI installer). To create a new MySQL user and to set the access policies to the
database, click here. |
||||||||||||||||||||
| Open database | - | Ctrl+O | Open a MetaQSAR database that can be a file or an ODBC data source. | |||||||||||||||||||||
| Close | - | - | Close MetaQSAR window without disconnecting the database. if you re-open MetaQSAR (Tools
|
|||||||||||||||||||||
| Edit | Expert mode | - | - | Checking this menu item, you enable the capability to edit all data. By default, MetaQSAR is protected from changes in order to avoid accidental modifications with the exception of the possibility to add new metabolic reactions. | ||||||||||||||||||||
| Tools | Calculate fingerprints | All | - | Calculate the fingerprints of all molecules
(substrates and products) in the database. When you perform this
calculation for the first time, the Molecules table of the
current database is modified adding three columns with a length of 255
characters (FpSim1, FpSim2 and FpSim3) in which the
fingerprints are stored in
Base64 format. So, to obtain the original fingerprint in binary
format, you must join the three fields (FpSim1 + FpSim2 +
FpSim3) and decode the resulting string using the Base64
scheme (the size of the fingerprint is 3736 bits, 467 bytes). These
fingerprints are employed to search molecules by similarity (see
Search tab). When you select this menu item, a progress bar is shown at the bottom of MetaQSAR window and you can stop the calculation by clicking the Abort button. |
||||||||||||||||||||
| Update | - | By this function, you can calculate the fingerprints only for the molecules for which the data is missing. This feature is useful when you added new molecules to the database and you need to calculate the fingerprints only for these ones. | ||||||||||||||||||||||
| Help | Manual | - | Ctrl+H | Show this manual. | ||||||||||||||||||||
| About | - | - | Show the copyright message. | |||||||||||||||||||||
Before to start to edit the metabolic reactions, you must follow these steps to prepare the data:
If you want to mange also the bibliographic data, you should:
Now you are ready to operate MetaQSAR. In the main window, there are five tabs allowing you to choose the different operating modes of the tool:
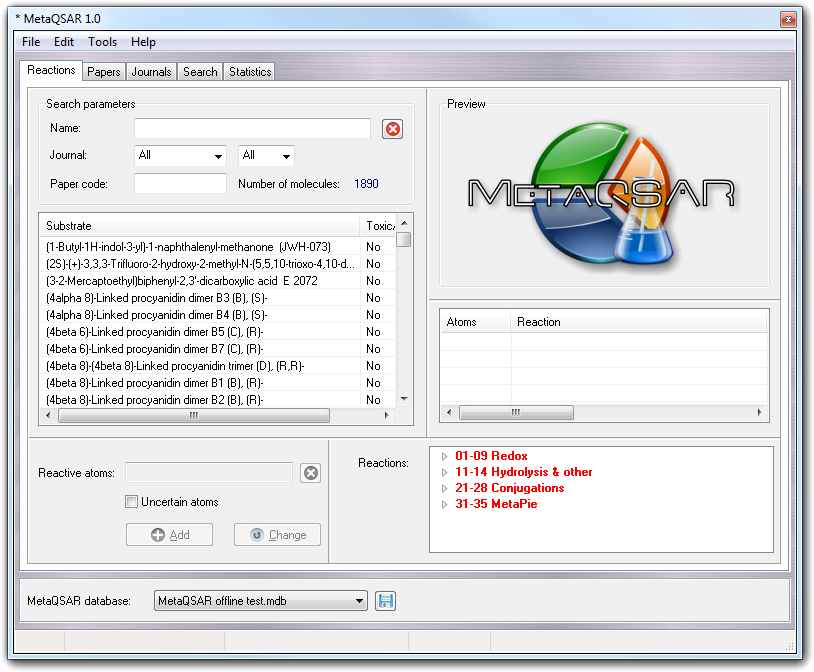
In this tab, you can manage the reactions linking them to the substrate and classifying them according to the MetaQSAR rules.
This tab allows the bibliographic data to be managed. In particular, you can add new papers and link metabolic substrates/products to their bibliographic source.
In this tab, you can edit the publishers and the journals used to classify the papers.
MetaQSAR is not only a tool for data entry, but includes also features to retrieve data with some capabilities to analyze it. In particular, in this tab, you can search substrates/products by structural similarity with a query molecule, or by molecular properties.
This tab shows statistics on the database.
In this tab, you can indicate not only which metabolic reactions can give each substrate but also which atoms are involved.
To input a reaction into the database, you must follow these steps:
You can use also the wildcards for more complex searches, but you must remember that SQL wildcards differs from DOS/Linux ones:
SQL DOS/Windows Linux Description % * * A substitute for zero or more characters. _ ? ? A substitute for a single character. [charlist] N/A [charlist] Sets and ranges of characters to match.
WARNING:
Linux have a different syntax: the characters must be comma separated.
e.g.
Linux: [a,b,c]
SQL: [abc]
The syntax for character ranges is the same: [a-c]
[^charlist] or
[!charlist]
N/A [!charlist] Matches only a character NOT specified within the brackets. The syntax differences are the same explained above.
The search by Journal or Paper code works properly only if each molecule is correctly linked to its paper (see Papers tab).
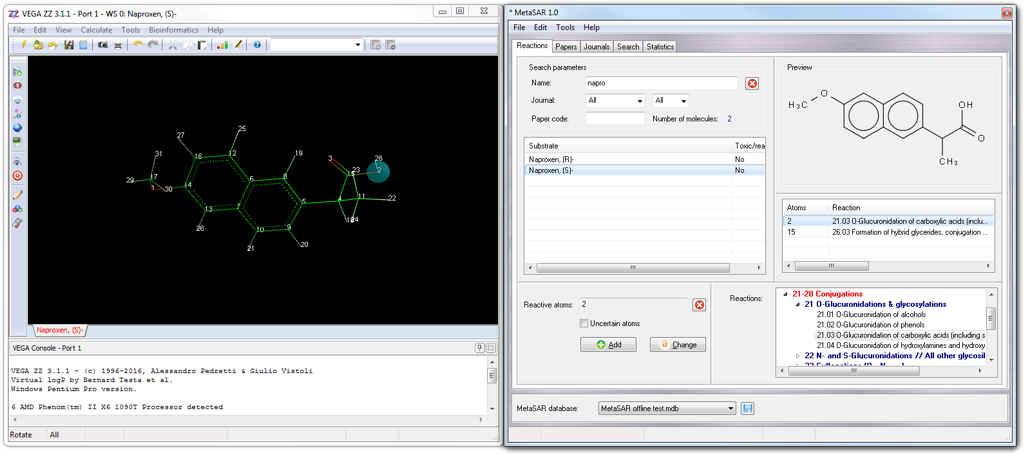
When you click the substrate name in the result list with the left mouse button, both 2D and 3D structures are shown respectively in MetaQSAR (Preview box) and VEGA ZZ windows. If you show the context menu of Preview box (click with the right mouse button), you can copy the 2D sketch of the molecule (Copy menu item) to the clipboard, copy the SMILES string to the clipboard (Copy SMILES) and print the 2D sketch.
More advanced searches and operations can be performed showing the context menu of the substrate list:
Item levels
Description 1 2 Find molecules with reactions Search for molecules whose metabolic reactions are already inserted in the database. without reactions Search for molecules whose metabolic reactions are not yet inserted in the database. with reaction notes Search for molecules whose metabolic reactions are already inserted with notes in the specific field. Edit - Enable the edit mode: by default this feature is disabled to avoid accidental changes in the database. When you enable it, Delete menu item becomes active and the background color of the substrate list changes from white to yellow. Moreover, clicking a substrate previously selected, you can rename it and change the Toxic/reactive flag. This special flag is useful to indicate if product is a toxic and/or reactive species.
Delete - Delete the substrate structure and all data about it (structure, molecular properties, reactions and links with the papers).
WARNING:
Delete substrates only by MetaQSAR and not by Database explorer, because only the former guarantees the data integrity, removing the cross-references in other tables.
Selecting a substrate with metabolic reactions, the ID of the reactive atoms (Atoms column), the full reaction description (Raection), the metabolic generation (Gen.), the involved enzyme (Enzyme), the alternative enzyme that can catalyze the reaction (Alt. enzyme), a flag indicating if the reaction gives toxic products (Toxic prod.) and the notes (Notes) are shown in the list below the Preview box. If you click a reaction in this box by left mouse button, the involved atoms are highlighted in VEGA ZZ main window as small transparent spheres coloured by dark green and besides, the tree of the Reactions box is automatically expanded in order to show you the main class and the class in which the reaction is grouped.
If you have to remove a reaction from a substrate, you must select the reaction clicking it and in the context menu, choose Delete. This operation is possible only in edit mode.
In both lists of substrates and reactions, as well as for all MetaQSAR lists, you can change the sorting mode clicking the column header with the left mouse button. For example, if if you click the Enzyme column for the first time, the list is sorted in ascending order by enzyme, but if you click it for the second time, it is sorted in descending order.
In this tab, you can manage the bibliographic resources adding new papers or editing them and linking substrates and products to their sources in which they are cited.
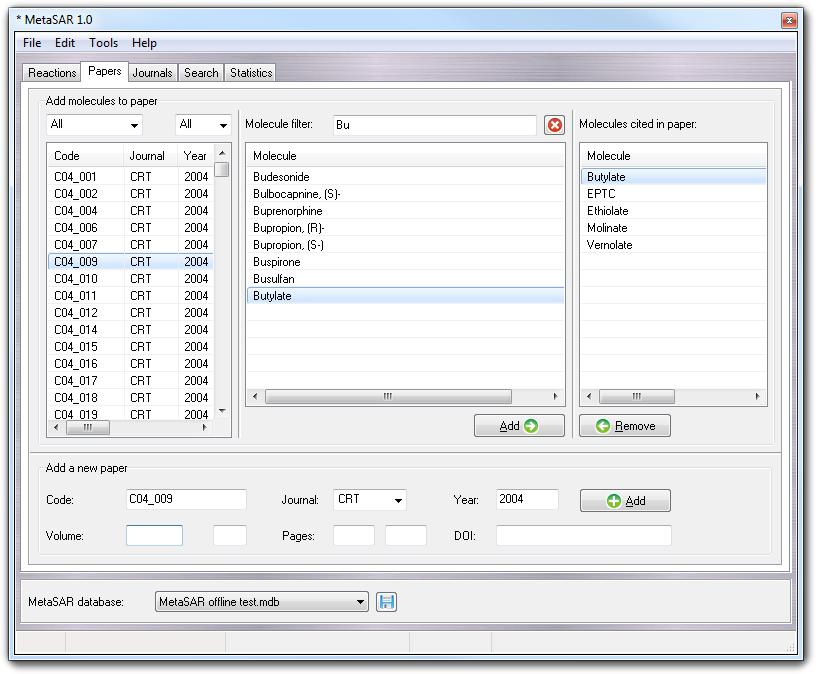
Here you can add new articles, filling the fields and clicking Add button in Add new paper box at the bottom of the window. The Journal combo-box is automatically updated with the data inserted in the Journals tab and only the short title is shown. Take care to define the paper code (Code field): 1) the maximum length of the code is 16 characters; 2) you can use your own convention, but you must use always the same. More in detail, in this example the papers are encoded as CYY_NNN where C is the journal ID (e.g. C = Chem. Res. Toxicol., D = Drug Metab. Dispos, X = Xenobiotica), YY is the publication year (e.g. 06 for 2006) and NNN is the progressive number.
All papers in the database are shown in the list at the left on the window and to simplify the view, you can filter them using the two pull-down menu at the top of the list. So, you can filter the paper by journal and/or year respectively with the leftmost and rightmost gadget.
The context menu of the paper list includes the features shown in the following table:
|
Item levels |
Description | |
| 1 | 2 | |
| Find papers | Orphan | Filter the papers not yet linked to any molecule. |
| View | - | Show the paper of the selected item. This function works only if DOI (Digital Object Identifier) is present and you have the subscription if the journal is not open access. |
| Edit | - | Enable/disable the edit mode. When you enable the edit mode, you can change all data of the selected item, just clicking them in the list. Moreover, you can also delete the whole record (see Delete item). |
| Delete | - | Remove the selected paper. This menu item is active only if the edit mode is enabled. |
| Refresh | - | Refresh the list of the papers. This function is needed only if another user changes the database, desynchronizing the list contents with the database. |
If you have to link one or more substrates/products to a paper, you must follow these steps:
Item levels
Description 1 2 Find molecules Orphan Filter the molecules not yet linked to any paper. Edit - Enable/disable the edit mode. When you enable the edit mode, you can rename and delete the selected molecule (see Delete item). Delete - Remove the selected molecule. This menu item is active only if the edit mode is enabled.
Item levels
Description 1 2 Edit - Enable/disable the edit mode. When you enable the edit mode, you can rename and delete the selected molecule (see Delete item). Delete - Remove the selected molecule. This menu item is active only if the edit mode is enabled.
WARNING:
Don't use the Delete item of the context menu (it's enabled only in edit mode) because it delete the molecule and not the link !
This tab allows the publishers and the journals to be managed, data that is used for the right classification of the papers (see Papers tab).
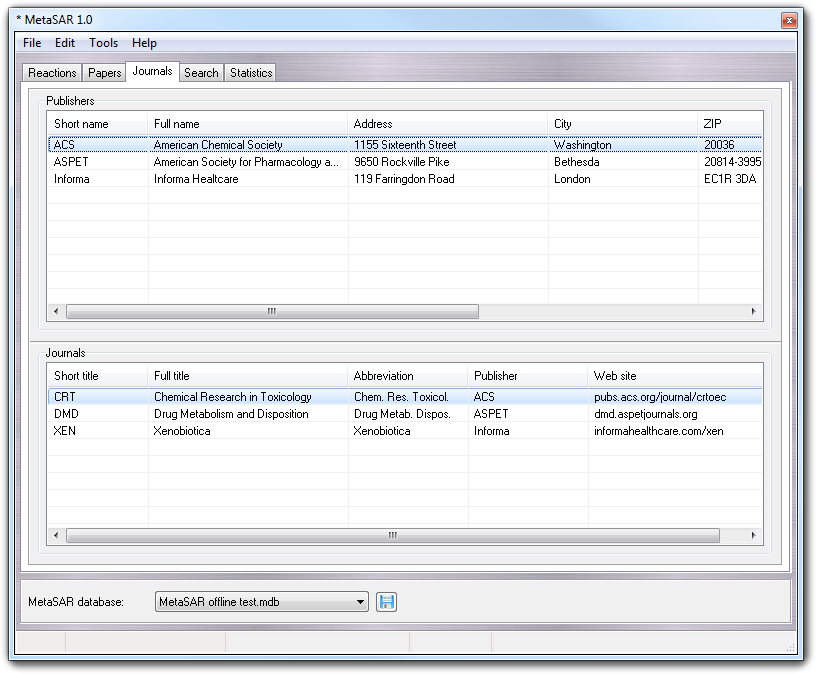
To operate this tab, you must use the context menu (it's the same for both Publishers and Journals lists) whose functions are shown in the following table:
|
Item levels |
Description | |
| 1 | 2 | |
| Open Web site | - | Show the Web site of the selected publisher or journal, if the Web site field is not empty. Remember that when you edit this field, you don't need to add http:// or https:// URL prefix. |
| Edit | - | Enable/disable the edit mode. When you enable the edit mode, you can add a new item and edit or delete a previously added one. |
| Add | - | Add a new empty item (function enabled only in edit mode). To edit the empty fields of the new item, just click them with the left mouse button. |
| Delete | - | Remove the selected item (function enabled only in edit mode). |
| Refresh | - | Refresh the list. As explained above, this function is useful if another user changes the database at the same time. |
To add a new publisher or journal you must follow these steps:
MetaQSAR includes some tools to retrieve and analyze the metabolic data stored in the database. In particular, in this tab, you can search for substrates/products by structural similarity with a query molecule, or by molecular properties.
12.8.2.4.1 Search by similarity
Before to run a similarity search, you must calculate or update the
fingerprints of all molecules included in the database, selecting Calculate
![]() Fingerprints in the main menu (for more details,
click here).
Fingerprints in the main menu (for more details,
click here).
Choosing By structure tab, you can search for substrates and related reactions, having a Tanimoto index greater than the specified value (see Similarity field). The input structure must be in SMILES format (see SMILES field) and can be edited typing manually the SMILES whose 2D sketch is shown in real time in the Query structure box. If your knowledge of SMILES language is poor, you can build or download the query molecule in VEGA ZZ, using one of the tools included in the program (2D, 3D, IUPAC editors, optical structure recognition, PubChem downloader, etc), after that you can click the Get button to transfer the SMILES string from the current workspace to MetaQSAR.
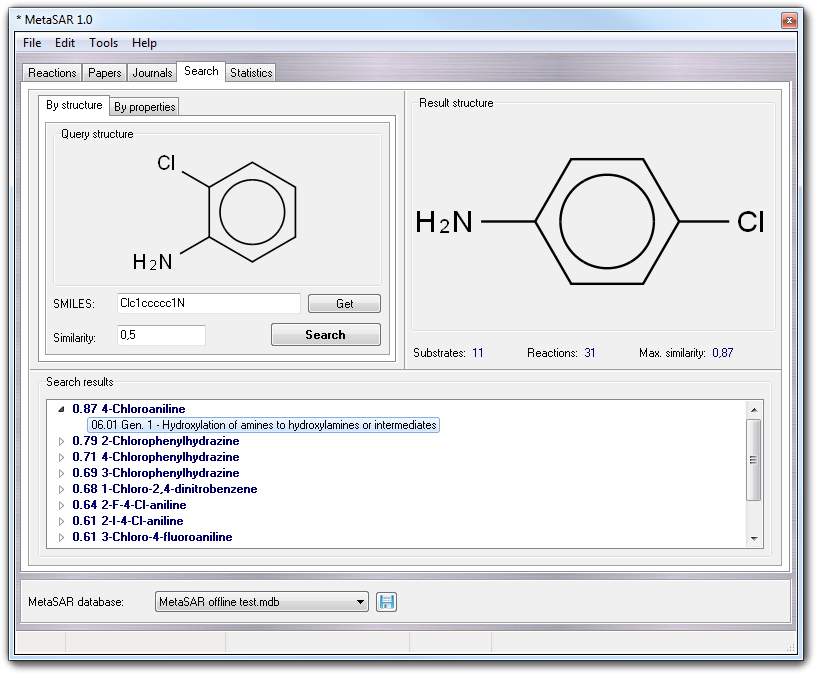
Clicking the Start button, the search begins and since the fingerprints are pre-calculated, is usually fast, nevertheless it can be stopped in any time clicking the Abort button shown in the status bar at the bottom of the window. The results are shown in Search result list: each line consists of the similarity index and the name of the substrate. Selecting a line, the substrate is shown as 2D sketch in Result structure box and in 3D in VEGA ZZ main window. Clicking the small triangle at the beginning of each line, you can show the reactions given by the substrate as code, generation number and description according to the MetaQSAR classification of the metabolic reactions. When you click the reaction, the involved atoms are highlighted in VEGA ZZ main window. The context menu of this output list includes interesting features as shown in the following table:
|
Menu item |
Description |
| Collapse | Collapse/expand the selected substrate to un/show the given metabolic reactions. |
| Expand | |
| Collapse all | Collapse/expand all substrates to un/show the given metabolic reactions. |
| Expand all | |
| Export to Excel | Export the results to Microsoft Excel. |
When you add a substrate to MetaQSAR, several molecular properties are calculated which can be used to perform searches. In By structure tab, you can build queries based on molecular properties adding the conditions in the table.
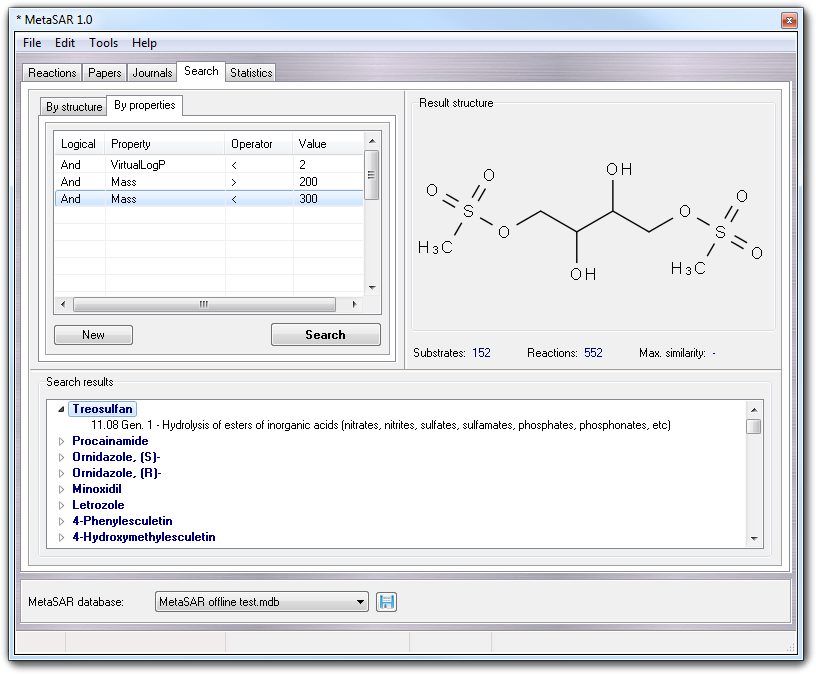
In each line of the table, you can add the Logical operator (and, or) , the molecular Property chosen by a pull-down menu, a mathematical Operator and the conditional Value. For a complete list of the properties, you can see the structure of the Molecules table (only the numerical properties are taken in account). If you add custom properties in Molecules table, they are added automatically to the pull-down menu.
To start the search, you must click the Search button, while to reset the query you can click the New button.
In the example shown in the above picture, 152 substrates giving 552 metabolic reactions are found according to a VirtualLogP value less than 2 and a mass value between 200 and 300 Daltons. As for the search by similarity, the results are shown in Search results box.
This tab shows interesting statistical values of all data included in the database.
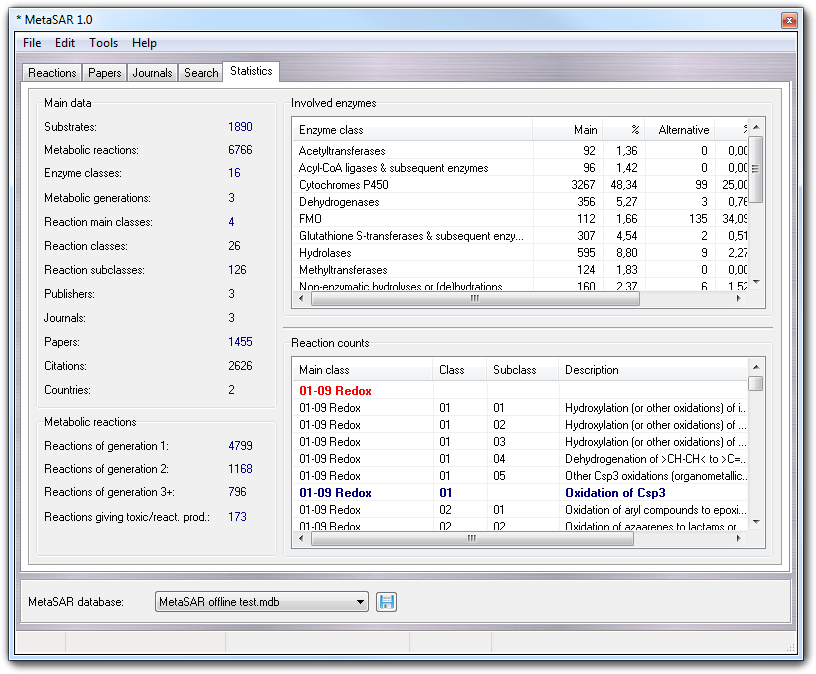
In the window, there are four sections summarizing the statistics:
This part of the window shows generic statistics such as the number of substrates, metabolic reactions, enzyme classes, metabolic generations, reaction main classes, reaction classes, reaction subclasses, publishers, journals, papers, citations and publisher countries.
In this section, you can find the counts of the reactions according to metabolic generation (1, 2 and 3+) in which they are involved. Besides, the number of reactions giving toxic and/or reactive products is also shown.
This list shows the statistics on the enzyme classes involved in the metabolic reactions. In particular, for each class it is reported the number of reactions catalyzed as main and alternative enzyme, the relative percentages and the total amount of reactions with its percentage (main + alternative).
Here the statistics on main classes, classes and subclasses of metabolic reactions are shown. More in detail, for each main class (in red), class (in dark-blue) and subclass (in black) is reported the number of metabolic reactions and its percentage.
The context menu of this tab allows you to refresh the data (Refresh item) and export the statistics to Microsoft Excel (Export to Excel item) for a better analysis.
MetaQSAR is a relational database containing both standard VEGA ZZ tables and specific ones to manage the metabolic reactions. The database is summarized in the following scheme in which the red arrows show the relationships between the tables.
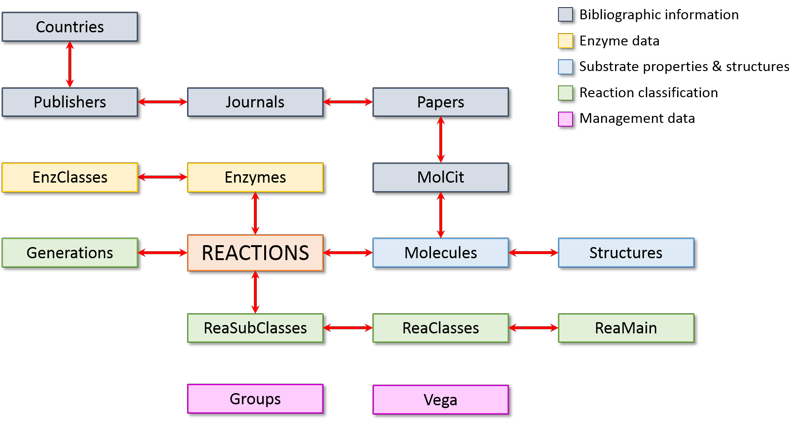
As shown in this chart, the most important table is reactions that includes the information to classify the metabolic reaction. Different colours are used to group tables including homogeneous data tables: grey for the bibliographic information, yellow for the enzymatic data, light blue for the substrate properties and their structures (1D, 2D and 3D), green for the reaction classification and pink for the management data.
Here, for each table, it is shown a short description, the fields, how they are defined (according to the SQL data types), the relationships between the tables and the default data included when an empty database is created:
Field Definition Description CountryID INTEGER Primary key (autoincrement). Country VARCHAR(50) Country name. Compact VARCHAR(20) Short name of the country. By default, in an empty MetaQSAR database are available two countries:
- United States of America (USA).
- United Kingdom (UK).
Field Definition Description ClassID INTEGER Primary key (autoincrement). Class VARCHAR(20) Enzyme class name. EC VARCHAR(8) EC classification code. MetaQSAR includes five main enzyme classes that can be changed and/or expanded by the user:
- Oxidoreductase
- Hydrolases
- Transferases
- Ligases
- Any
Field Definition Description EnzymeID INTEGER Primary key (autoincrement). Enzyme VARCHAR(100) Enzyme description. By default, MetaQSAR includes 16 enzyme classes:
- Cytochromes P450
- Dehydrogenases
- FMO
- XO, AO
- Peroxidases
- Other reductases
- Other oxidoreductases or autooxidations
- Hydrolases
- UDP-Glucuronosyltransferases
- Sulfotransferases
- Glutathione S-transferases & subsequent enzymes/reactions
- Acetyltransferases
- Acyl-CoA ligases & subsequent enzymes
- Methyltransferases
- Other transferases or non-enzymatic conjugations
- Non-enzymatic hydrolyses or (de)hydrations
Field Definition Description GenerationID INTEGER Primary key (autoincrement). Generation VARCHAR(4) Generation description (1, 2, 3+). Only three generations of metabolic products are considered by MetaQSAR, because the number of metabolites of 4th or more generation is negligible:
- First generation (1)
- Second generation (2)
- Third generation or more (3+)
Field Definition Description ID INTEGER Primary key (autoincrement). Group VARCHAR(50) Name of the user group. Date DATE Creation date of the record. Time TIME Creation time of the record.
Field Definition Description TitleID INTEGER Primary key (autoincrement). Title VARCHAR(10) Short name for paper encoding (e.g. CRT, DMD and XEN). FullTitle VARCHAR(128) Full name of the journal. Abbreviation VARCHAR(50) Journal title abbreviation. EditorID INTEGER Editor ID (see publishers table). URL VARCHAR(50) Web site URL of the journal. By default, MetaQSAR includes three of the most important journal on metabolism, but others can be added by the user:
- Chemical Research in Toxicology
- Drug Metabolism and Disposition
- Xenobiotica
Field Definition Description ID INTEGER Primary key (autoincrement). MolID INTEGER ID of the substrate involved in the reaction shown in the paper (see molecules table). PaperID INTEGER ID of the paper including the reaction that involves the substrate (see papers table). Date DATE Creation date of the record. Time TIME Creation time of the record.
Field Definition Description ID INTEGER Primary key (autoincrement). Name VARCHAR(255) Molecule name. IUPAC VARCHAR(255) IUPAC name. Formula VARCHAR(50) Chemical formula. Smiles VARCHAR(255) Smiles structure. Inchi VARCHAR(255) InChI structure. InchiKey VARCHAR(28) InChI key. GroupID INTEGER Group ID (not used). Cas VARCHAR(50) Chemical Abstract code. Code VARCHAR(50) Auxiliary code (not used). Angles INTEGER Number of bond angles. Atoms INTEGER Number of atoms. Bonds INTEGER Number of bonds. Charge INTEGER Formal charge. ChiralAtms INTEGER Number of chiral atoms. Dipole REAL Dipole moment. ExBnds INTEGER Number of bonds giving geometric isomers (E/Z). FlexTorsions INTEGER Number of flexible dihedral angles (torsions). FuncGroups VARCHAR(100) List of the functional groups. This string has the following format: NUM_1 GRP_1 NUM_2 GRP_2 ... NUM_N GRP_Nwhere NUM is the number of functional groups of kind GRP. The functional groups are detected by the GROUPS.tem ATDL template (see the Data directory), as shown in the following table:
Group Description COOH Carboxylic acid. COOR Ester. CHO Aldheyde. CON2 Urea. CON Amide. OCOO Carbonate. COCl Acyl chloride. COBr Acyl bromide. CNH Aldimine. CNR Imine. CO Ketone. OCN Cyanate. NCO Isocyanate. NCS Tiocyanate. CN Nitrile. N1 Primary amine. N2 Secondary amine. N3 Tertiary amine. N+ Ammonium salt. NP Aromatic planar nitrogen. NO3 Nitrate.
Group Description NO2 Nitrite. NNN Azide. NO Nitrose. NC Isocyanide. OH2 Water. OH Alchol. PhOH Phenol. OR2 Ether. 2O2 Peroxyde. SO3H Sulfonic acid. SO2 Sulfone. SO Sulfoxide. SH Thiol. SR2 Thioether. 2S2 Disulfide. PO4 Phosphate P3 Phospine. F Fluoride. Cl Chloride. Br Bromide. I Iodide.
Gyrrad REAL Gyration radius (Å). HbAcc INTEGER Number of H-bond acceptors. HbDon INTEGER Number of H-bond donors. HeavyAtoms INTEGER Number of heavy atoms. Impropers INTEGER Number of improper/pyramidal angles (out-of-plane). Lipole REAL Lipophilicity moment. Mass REAL Molecular weight (Daltons). MassMI REAL Monoisotopic mass (Daltons). Molecules INTEGER Number of molecules included in the record. This value is usually set to 1, but can be greater for salt, complexes, etc. Ovality INTEGER Ovality is a form factor: it's close to 1 for spherical molecules. Psa REAL Polar surface area (Å2). Rings INTEGER Number of rings. Sas REAL Solvent accessible area (Å2). Sav REAL Solvent accessible volume (Å2). Sdiam REAL Surface diameter: diameter of the equivalent sphere with the same surface of the molecule (Å). Surface REAL Van der Waals surface area (Å2). Torsions INTEGER Number of dihedral angles (torsions). Vdiam REAL Volume diameter: diameter of the equivalent sphere with the same volume of the molecule (Å). VirtualLogP REAL Log P calculated with Bernard Testa's method. Volume REAL Volume (Å3). Toxic INTEGER Toxicity flag: it can be 0 or 1 respectively for non-toxic or toxic molecules. Date DATE Creation date of the record. Time TIME Creation time of the record. FpSim1 VARCHAR(255) These columns are optional and are added automatically when you calculate the fingerprints of the substrates/products included in the current database (see Tools Calculate fingerprints).
FpSim2 VARCHAR(255) FpSim3 VARCHAR(255)
Field Definition Description PaperID INTEGER Primary key (autoincrement). Paper_Code VARCHAR(16) Paper ID. The convention used to encode the paper ID is: CYY_NNNwhere C is the journal ID (e.g. C = Chem. Res. Toxicol., D = Drug Metab. Dispos, X = Xenobiotica), YY is the publication year (e.g. 06 for 2006) and NNN is the progressive number.
JournaID INTEGER Journal ID (see journals table). Year INTEGER Publication year. Volume INTEGER Volume. Issue VARCHAR(8) Issue. FirstPage INTEGER First page. LastPage INTEGER Last page. Doi VARCHAR(32) Digital object identifier (DOI). Date DATE Creation date of the record. Time TIME Creation time of the record.
Field Definition Description EditorID INTEGER Primary key (autoincrement). Editor VARCHAR(50) Short name of the editor. FullEditor VARCHAR(128) Full name of the editor. Address VARCHAR(128) Address. City VARCHAR(50) City. Postal_code VARCHAR(32) Postal code. State VARCHAR(50) State. CountryID INTEGER ID of the country (see countries table). URL VARCHAR(50) Web site URL (without http://).
Field Definition Description ID INTEGER Primary key (autoincrement). MainID INTEGER ID of the main class in which it's included (see reamain table). ClassCode VARCHAR(8) Alphanumerical class code for easy class identification. Description VARCHAR(128) Full description of the reaction class.
The 21 reaction classes are:
Num. Main class Class code Description 1 Redox 01 Oxidation of Csp3 2 02 Oxidation of Csp2 & Csp 3 03 -CHOH <-> >C=O -> -COOH 4 04 Various redox reactions of carbon atoms 5 05 Redox reactions of R3N 6 06 Oxidation of >NH, >NOH and -N=O // Reduction of -NO2, -N=O, >NOH, etc. 7 07 Oxidation to quinones or analogs // Reduction of quinones and analogs 8 08 Oxidation and reduction of S atoms 9 09 Redox reactions of other atoms 10 Hydrolysis & other 11 Hydrolysis of esters, lactones and inorganic esters 11 12 Hydrolysis of amides, lactams and peptides 12 13 Epoxide hydration 13 14 Other hydrolysis/hydration reactions // Non-enzymatic eliminations and rearrangements 14 Conjugations 21 O-Glucuronidations & glycosylations 15 22 N- and S-Glucuronidations // All other glycosilations 16 23 Sulfonations (O-, N-, ...) 17 24 GSH & RSH conjugations + sequels // GSH-mediated reductions 18 25 Acetylations & acylations 19 26 CoASH-Ligation followed by amino acid conjugations or other sequels 20 27 Methylations (O-, N-, S-) 21 28 Other conjugations (PO4, CO2, ...) // Transaminations
Field Definition Description ID INTEGER Primary key (autoincrement). MolID INTEGER ID of the molecule involved in the reaction (see molecules table). ReaSubClassID INTEGER ID of the sub class of reactions in which the reaction is included (see reasubclasses table). GenerationID INTEGER ID of the product generation (see generations table). EnzymeID1 INTEGER ID of the main enzyme catalyzing the reaction (see enzymes table). EnzymeID2 INTEGER ID of the alternative enzyme catalyzing the reaction (see enzymes table). Atoms VARCHAR(50) Comma-separated list of the atoms involved in the reaction. For each atom is reported its ID of corresponding 3D structure (see structures table). For uncertain reactive atoms, the ID is followed by a question mark (?). ProdID INTEGER ID of the molecule produced by the reaction (see molecules table, not yet used). ProdActive INTEGER Boolean flag indicating if the product is active or nor (no more used, kept for MetaPies compatibility). ProdReact INTEGER Boolean flag indicating if the product is reactive/toxic. Notes VARCHAR(64) Field for generic notes. Date DATE Creation date of the record. Time TIME Creation time of the record.
Field Definition Description ID INTEGER Primary key (autoincrement). ShortDesc VARCHAR(32) Short description (used in the reaction tree of MetaQSAR plug-in). Description VARCHAR(80) Full description. The following table shows the default entries:
ID Short description Full description 1 01-09 Redox Redox reactions 2 11-14 Hydrolysis & other Reactions of hydrolysis and other non-redox functionalizations 3 21-28 Conjugations Conjugation reactions
Field Definition Description ID INTEGER Primary key (autoincrement). ClassID INTEGER Reaction class ID including this subclass (see reaclasses table). MetaPieID INTEGER MetaPiesID (no more used). Description VARCHAR(200) Reaction class description. By default, a MetaQSAR database includes 101 reaction subclasses in this table as shown below:
Num. Main class Class Subclass code Description 1 Redox 01 Oxidation of Csp3 01 Hydroxylation (or other oxidations) of isolated Csp3 2 02 Hydroxylation (or other oxidations) of C alpha to an unsaturated system (>C=CC=O, -C±N, aryl) 3 03 Hydroxylation (or other oxidations) of Csp3 carrying an heteroatom (N, O, S, halo) (including subsequent dealkylation, deamination or dehalogenation) 4 04 Dehydrogenation of >CH-CH< to >C=CCH-N< to >C=N- (incl. >C=N+<) 5 05 Other Csp3 oxidations (organometallic dealkylation, C-C cleavage, etc) 6 02 Oxidation of Csp2 & Csp 01 Oxidation of aryl compounds to epoxides, phenols or other metabolites 7 02 Oxidation of azaarenes to lactams or other metabolites 8 03 Oxygenation of >C=C< bonds to epoxides or other metabolites 9 04 Oxygenation of -C-C±C-H and -C-C±C-C- bonds 10 03 -CHOH <-> >C=O -> -COOH 01 Dehydrogenation of -CH2OH groups to -CHO and of >CHOH to >C=O 11 02 Hydrogenation of -CHO to -CH2OH and of >C=O to >CHOH 12 03 Oxidation of -CHO to -COOH 13 04 Reduction of arene and alkene epoxides 14 04 Various redox reactions of carbon atoms 01 Oxidative decarboxylation 15 02 Reductive dehalogenations 16 03 Reduction of arene and alkene epoxides 17 04 Other C reductions, e.g. of >C=C< to -CH2-CH2- 18 05 Redox reactions of R3N 01 Oxidation of tertiary alkylamines and heterocyclic amines to N-oxides or other metabolites 19 02 Oxidation of tertiary arylamines, azaarenes and azo compounds to N-oxides oxides or other metabolites 20 03 Reduction of N-oxides 21 06 Oxidation of >NH, >NOH and -N=O // Reduction of -NO2, -N=O, >NOH, etc. 01 Hydroxylation of amines to hydroxylamines or intermediates 22 02 Hydroxylation of amides to hydroxylamides 23 03 Oxidation of primary hydroxylamines to nitroso compounds or oximes (incl. spontaneous dismutation), then to nitro compounds 24 04 Reduction of hydroxylamines and hydroxylamides (incl. spontaneous dismutation) 25 05 Reduction of nitroso compounds and oximes to hydroxylamines 26 06 Reduction of nitro compounds to nitroso compounds 27 07 Other N-oxidations (1,4-dihydropyridines, etc) 28 08 Other N-reductions (e.g. azo compounds to hydrazines, hydrazines to amines, reductive N-rings opening) 29 07 Oxidation to quinones or analogs // Reduction of quinones and analogs 01 Oxidation of diphenols to quinones 30 02 Oxidation of amino- and amido-phenols to quinoneimines or quinoneimides, resp. 31 03 Oxidation of cresols and analogs to quinonemethides 32 04 Other oxidations of phenols and amines (dimerization, quinone-like metabolites, etc) 33 05 Reduction of quinones and analogs 34 08 Oxidation and reduction of S atoms 01 Oxidation of thiols to sulfenic acids or disulfides 35 02 Oxygenation of sulfenic acids to sulfinic acids, and of sulfinic acids to sulfonic acids 36 03 Oxygenation of sulfides to sulfoxides, and of sulfoxides to sulfones 37 04 Oxygenation of thiones (>C=S) or thioamides to sulfines, and of sulfines to sulfenes 38 05 Oxidative desulfurations of >C=S to ketones, and of -P=S to -P=O groups 39 06 S-Oxygenations of disulfides, thiosulfinates (-SO-S-), alpha-disulfoxides (-SO-SO-) and thiosulfonates (-SO2-S-) 40 07 Reduction of disulfides to thiols 41 08 Reduction of sulfoxides to sulfides 42 09 Other S-reductions 43 09 Redox reactions of other atoms 01 Oxidation of silicon, phosphorus, arsenic and other atoms 44 02 Reduction of Se, P, Hg, As and other atoms 45 Hydrolysis & other 11 Hydrolysis of esters, lactones and inorganic esters 01 Hydrolysis of alkyl esters 46 02 Hydrolysis of aryl esters 47 03 Hydrolysis of anionic and cationic esters 48 04 Hydrolysis of linear and cyclic carbamates (>N-CO-OR') and carbonates (RO-CO-OR') 49 05 Hydrolysis of acyl ß-glucuronides or other acylglycosides 50 06 Reversible hydrolytic opening of lactone rings 51 07 Hydrolysis of thioesters (RCO-SR' and RCS-SR') and thiolactones 52 08 Hydrolysis of esters of inorganic acids (nitrates, nitrites, sulfates, sulfamates, phosphates, phosphonates, etc) 53 12 Hydrolysis of amides, lactams and peptides 01 Hydrolysis of alkyl and aryl amides [alkyl-CO-N< and aryl-CO-N<] 54 02 Hydrolysis of anilides [aryl-N-CO-C~], hydrazides [-CO-NHNN-CO-N<] 55 03 Hydrolysis of lactams, cyclic imides and cyclic ureides 56 04 Hydrolysis of peptide bonds 57 13 Epoxide hydration 01 Hydration of arene and alkene oxides 58 14 Other hydrolysis/hydration reactions // Non-enzymatic eliminations and rearrangements 01 Hydrolysis of glucuronides and other glycosides (including N- and S-glycosides) 59 02 Other ether hydrolyses (benzhydryl ethers, acetals, etc) 60 03 Hydrolytic cleavage of C=N bonds (in imines, hydrazones, imidates, amidines, oximes, oximines, isocyanates, etc), and of C±N bonds (nitriles). Double bond hydration 61 04 Hydrolysis of linear Mannich bases (N-, O- and S-Mannich bases) and cyclic Mannich bases (imidazolidines, oxazolidines, etc) 62 05 Hydrolytic opening of other ring systems (1,2-oxazoles, etc) 63 06 Hydrolytic dehalogenations 64 07 Substitution reactions (by H2O, halide, etc) in complexes of Pt or other metals (with elimination of halide or other ligands) 65 08 Bond order increases by elimination of H2O or RSH or other Nu-H 66 09 Cyclizations by intramolecular nucleophilic substitution (with elimination of amine, phenol, halide or H2O) 67 10 Any other non-redox, non-conjugation reaction 68 Conjugations 21 O-Glucuronidations & glycosylations 01 O-Glucuronidation of alcohols 69 02 O-Glucuronidation of phenols 70 03 O-Glucuronidation of carboxylic acids (including subsequent rearrangements) 71 04 O-Glucuronidation of hydroxylamines and hydroxylamides 72 22 N- and S-Glucuronidations // All other glycosilations 01 N-Glucuronidation of linear and cyclic amines (including =N- in azaarenes) 73 02 N-Glucuronidation of amides 74 03 S-Glucuronidation of thiols and thioacids, C-Glucuronidation of acidic enols 75 04 Any conjugations with glucose or other sugars 76 23 Sulfonations (O-, N-, ...) 01 O-Sulfonation of phenols 77 02 O-Sulfonation of alcohols 78 03 Other reactions (O-sulfonation of hydroxylamines, N-sulfonation of amines, etc) 79 24 GSH & RSH conjugations + sequels // GSH-mediated reductions 01 Nucleophilic additions of glutathione (to a,ß-unsaturated carbonyls, quinones and analogues, isocyanates and isothiocyanates, epoxides, etc) 90 02 Reactions of glutathione addition-elimination (at C-X groups, acyl halides, halogenated olefins, etc) 81 03 Metabolic processing of glutathione conjugates up to thiols 82 04 Conjugation of glutathione with Hg, As, Pt, etc, compounds 83 05 Conjugations with other thiols (Cys, N-Ac-Cys, etc) 84 06 Reductions following glutathione conjugations 85 07 Radical scavenging by glutathione and other thiols 86 25 Acetylations & acylations 01 N-Acetylation of aromatic amines 87 02 N-Acetylation of hydrazines and hydrazides 88 03 Other acetylations (N-acetylation of alkylamines, O-acetylation, etc) 89 04 Reactions of acylation (formylation, formation of fatty acyl esters, etc) 90 26 CoASH-Ligation followed by amino acid conjugations or other sequels 01 Conjugation with glycine, glutamic acid, taurine and other amino acids or short peptides 91 02 Conjugation with carnitine 92 03 Formation of hybrid glycerides, conjugation with cholesterol or other sterols 93 04 Unidirectional chiral inversion of profens and analogues 94 05 Chain elongation by 2C, beta- or alpha- oxidation (loss of 2C or 1C, resp.), other sequels 95 27 Methylations (O-, N-, S-) 01 O-Methylation of catechols and other hydroxy groups 96 02 N-Methylation of exocyclic and endocyclic amino groups (including =N- in azaarenes) 97 03 S-Methylation of thiols 98 04 Methylation of metals and metalloids (Hg, As, etc) 99 28 Other conjugations (PO4, CO2, ...) // Transaminations 01 Reactions of phosphorylation 100 02 Non-enzymatic formation of hydrazones, binding of CO2 to form carbamates 101 03 Other reactions of conjugation, transaminations
Field Definition Description ID INTEGER Primary key (autoincrement). Structure BLOB Binary object including the 3D compressed structure in IFF/RIFF format.
Field Definition Description Key VARCHAR(16) Name of the data key. Value VARCHAR(32) Key value. Possible keys are:
Key Description 3DComp Compression algorithm used to store the 3D structures (see structures table). 3DFormat File format used to store the 3D structures (see structures table). MetaQSAR This optional key can assume the value of 1 if the database includes the MetaQSAR tables or 0 if not. It it's missing, is a normal VEGA ZZ database.