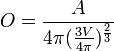3. Usage
Running the program without parameters, the list of the implemented options is shown:
VEGA 3.2.3 - (c) 1996-2023,
Alessandro Pedretti & Giulio Vistoli
Virtual logP by Bernard Testa et al.
Windows x64 (64 bit) version
Synopsis: vega INPUT ...
-o[OUT.PACK] -f[OUTPUT_FORMAT]
-p[FORCE_FIELD]
-s[POINTS] -g[RADIUS]
-c[TEMPLATE] -k[KEYWORDS]
-a[RES_NUM]
-d[DIELECTRIC] -e[MOLNUM]
-i[SHELL RAD SHAPE]-j[TORSIONS]
-l[MOLTYPE]
-m[KEYWORDS]
-q[METHOD]
-r[MODE]
-t[SECSTRUCT]
-v[CPUS] -x[MODE
(ID)]
-z[NTERM CTERM]
-0bhnuwy
0 -> ignore locale
settings of the decimal separator
a -> renumber residues starting from RES_NUM
b -> don't save the connectivity
c -> charge template (FORMAL, GASTEIGER,
...)
d -> dielectric constant for energy calculation
e -> molecule number for score calculation (0
= last)
f -> output format
g -> probe radius for SAS
h -> show this help
i -> solvate the molecule
j -> define the torsions (ALL, AUTODOCK, FLEX)
k -> keywords for InfoXML and MopInt
l -> add hydrogens (GEN, GENBO, NA, NABO, PROT, PROTBO)
m -> keywords for trajectory analysis
n -> normalize coordinates
o -> output file name
p -> define force field to apply
q -> fix the bond order (ALL, RINGS)
r -> remove hydrogens (ALL, APOLAR)
s -> point density for SAS
t -> change the protein secondary structure
u -> add the side chains to a protein
v -> number of CPUs (0 = all)
w -> remove waters
x -> list/extract molecule/s from a database (LIST; NAME name; NUM number)
y -> find the molecules in the assembly
z -> N-term and C-term capping for peptide (default: NONE
NONE)
INPUT formats:
Alchemy, AMMP, Arc, AutoDock 4 DLG, BioDock, CAR, CHARMM CRD, CIF, CML,
CML 2.0, CPMD XYZ, CRT, CHARMM DCD, Chem3D, ChemDraw CDX, ChemSol, CSSR,
EMPIRE, ESCHER NG, Fasta, GAMESS, Gaussian In/Out, GRAMM, Gromacs/Gromos
mol, Gromacs TRR, Gromacs XTC, HIN, IFF, InChI, LiGen pocket, MDL,
MDL V3000, Mol2, Mopac cartesian, Mopac Gaussian Z-matrix, Mopac internal,
MSF, NAMD binary, PDB, PDBA, PDBF, PDBL, PDBQT, PQR, PQRXML, PSFX, QMC,
Quanta CSR, RIFF, SDF, TINKER XYZ, XYZ, ZIP.
OUTput formats:
Calc: Info, InfoXML,
Score.
Map: BiosymSrf, ComfaFld, CsvIlm, CsvLogP, CsvMep, CsvSrf,
QuantaIlm, QuantaLogP, QuantaMep,
QuantaSrf.
Molecule: Alchemy, AMMP, Biosym, ChemSol, CIF, CML, CML2, CPMDXYZ, CRD,
CRT, CSSR, Fasta, GAMESS, GaussIn, Gromos, GromosNm, IFF,
InChI, InChIAux, InChIKey,
Indigo, MdlMol, MdlMol3, mmCIF,
Mol2, MopCar, MopInt, MSF, NamdBin, OldBiosym, PDB, PDB2,
PDBQ, PDBA, PDBF, PDBL, PDBNOTSTD, PDBQT, PQR, PQRXML,
PSFX, QMC,
RIFF, SMILES, SpilloRBS, VINA, XYZ.
Plot: BinPlt, CSV, QuantaPlt.
Trajectory: TrjDCD, TrjIFF, TrjMol2, TrjPDB, TrjTrr,
TrjXtc.
VRML: Vrml, VrmlPts, VrmlCpk,
VrmlSol.
PACKer formats:
bz2 (BZip2), gz (GZip), pp (PowerPacker), z (Z-Compress).
Score functions (-f Score -k):
Broto, Broto2, Broto3, Charmm, Charmm22, Charmm36, CVFF, Elect, ElectDD.
TRAJECTORY keywords (-m):
Angle A1 A2 A3, Dipole, Distance A1 A2, Extract F1 [F2], GyrRad, ILM,
LipoleBr, LipoleCr, Ovality, PlaneAng A1 A2 A3 A4 A5 A6, PSA, RMSD,
RMSDH, RMSDALN, RMSDALNH, RMSDSYMCOR, RMSDSYMCORH, Surface A1 ...,
SurfDia A1 ..., Torsion A1 A2 A3 A4, VlogP, VolDia, Volume.
Secondary structure keywords (-t):
AlphaHelix, LeftHelix, 310Helix, PiHelix, Beta, BetaAnti, BetaPar.
or
TOR=VALUE TOR=VALUE ...
where TOR is the torsion name (Phi, Psi, Omega) and VALUE is the
torsion value in degree.
Peptide capping keywords
(-z):
NTERM: NONE, H3N+, HCONH, H3CCONH
CTERM: NONE, O-, OH, OCH3, OC2H5
All parameters are optional with the exception of the input file name (INPUT).
This option allows to specify the input file names. VEGA recognizes
automatically the format of input files and the list of supported input
formats is shown running VEGA without arguments.
You can load more than one file at once with the same or different file formats to create
molecular assemblies. The calculation of connectivity is performed separately for each
file to prevent connectivity errors when the molecules are overlapped.
The Data Decompressor Engine allows to manage compressed files as
normal unpacked files without any
external data decompressor. VEGA supports the following compression formats:
| Format name | File extension |
| BZip2 | .bz2 |
| GZip | .gz |
| PowerPacker | .pp |
| Unix Un/compress | .Z |
VEGA can recognize .url files and open URLs specified as file names, downloading the molecules for you.
With this option, VEGA ignores the locale settings and writes always a dot (.) as decimal separator.
This option renumbers all residues starting from [RES_NUM]. If this value is not specified, VEGA starts from one. The residue renumbering is very useful when you create an assembly starting from two or more molecules.
This switch saves molecules without connectivity records when the output format can store this kind of information (e.g. PDB, PDBF, IFF). Many molecular packages interpret incorrectly the CONECT field in PDB files, therefore, to solve this problem, you can save the molecule without connectivity.
Currently, VEGA supports formal charges (formal keyword), atomic charges based on a fragment database (charmm22_char, charmm36_char, opls_char keywords) and atomic charges based on the Gasteiger-Marsili method (gasteiger keyword) . The Gasteiger-Marsili approach is based on a multi-step procedure:
The formal charges are correctly assigned only if all bonds have the right order (single, double and triple).
Use this option If you want to calculate the interaction energy (see -f[FORMAT] option) changing the default dielectric constant (1.0). Please note that the default value of dielectric constant is stored in the prefs file.
This is a compulsory parameter for the interaction energy evaluation (docking score evaluation, see -f[FORMAT] option). It is required to know which molecule (ligand) is considered to evaluate the interaction energy. You can specify 0 as molecule number to indicate the last molecule in the assembly.
WARNING:
the IFF/RIFF file format is the only one that is able to contain the molecule
number information. For this reason, it's impossible to select the ligand by
molecule number if you use assemblies (files containing more than one molecule)
in other formats. To skip the problem, you can build the assembly on-the-fly
specifying the ligand and the receptor as in the following example:
vega receptor.pdb ligand.pdb -f score -c gasteiger -k "CHARMM36 ELECT" -e 0 -o receptor-ligand.xml
Another solution is the use of -y options that enables the detection of molecules:
vega assembly.pdb -f score -c gasteiger -k BRORO -e 2 -o score.xml -y
With this parameter, you can create an output file in a specific file format. If -f is omitted, the default output format is PDB full standard (see PDB specifications) unpacked. OUT indicates the format and PACK is the optional compression method (bz2, gz, pp and z, see INPUT). This two keywords are case-insensitive.
| e.g. | -f CSSR | CSSR output without compression. | ||
| -f pdb.Z | PDB output with Unix compression. | |||
| -f xyz.bz2 | XYZ output with BZip2 compression. |
| Keyword | Description |
INFO |
Information about the molecule. |
| INFOXML | Same of above but the results are included in a XML file. |
| Score | Evaluation of interaction energy (molecular docking score). |
3.8.1.1 Information about the molecule
If you want more information about the input molecule, you can use -f INFO option. When you select this operation, VEGA shows many information: total number of atoms, number of heavy atoms, number of residues, number of molecules contained, number of water molecules, molecular weight, coordinates of geometric center, coordinates of mass center, approximative dimensions, total charge (calculated using the atomic charges), dipole, surface area, surface diameter, volume, volume diameter, ovality (only if the probe radius used for surface calculation is null, see -g option), Crippen's logP and lipole, Broto's logP and lipole, Virtual logP (available only in full release), predicted charge (only for proteins, it's calculated searching ionizable groups), aminoacidic charge (only for proteins, it's calculated at physiological pH on the basis of aminoacidic composition), aminoacidic or nucleotidic composition:
************************************ **** Information about molecule **** ************************************ Atoms..............: 48 Heavy atoms........: 25 Residues...........: 1 Molecules..........: 1 Waters.............: 0 Formula............: C19H23NO5 Molecular weight...: 345.384 Daltons Monoisotopic mass..: 345.157623 Daltons Geometry center....: 7.1076 3.6789 0.5790 Mass center........: 6.9492 3.5914 0.5256 Appx. dimensions...: 17.4088 10.7721 10.7163 Total charge.......: 0.0003 Dipole.............: 1.0292 Debye Surf. area (0.00)..: 383.3 Ų (ds=11.0 Å) Polar area (PSA)...: 50.6 Ų (apolar=332.7 Ų) Volume.............: 362.3 ų (dv=8.8 Å) Ovality............: 1.6 logP (Crippen).....: 1.9275 Lipole (Crippen)...: 0.4363 logP (Broto).......: 3.0390 Lipole (Broto).....: 0.4755 Virtual logP.......: 3.1402
Please note that the total number of atoms exceeds the MAXATMINFO key in prefs file, surface area, surface
diameter, volume, volume diameter, ovality and logP values are not shown.
If the molecule is a protein or a nucleic acid, the following data are shown:
...
Total charge.......: -23.0004 Predicted charge...: -24 Aminoacidic charge.: -24
Aminoacidic composition:
Res N. N. % Mass Mass % ==================================== ALA 46 6.29 3269.690 3.57 ARG 42 5.75 6618.506 7.22 ASN 29 3.97 3309.140 3.61 ASP 43 5.88 4921.520 5.37 CYS 18 2.46 1855.515 2.02 GLU 53 7.25 6789.680 7.41 GLN 46 6.29 5894.132 6.43 GLY 40 5.47 2282.165 2.49 HIS 26 3.56 3565.805 3.89 ILE 37 5.06 4186.861 4.57 LEU 86 11.76 9731.422 10.62 LYS 30 4.10 3875.539 4.23 MET 11 1.50 1443.115 1.57 PHE 25 3.42 3681.328 4.02 PRO 35 4.79 3401.088 3.71 SER 42 5.75 3657.370 3.99 THR 24 3.28 2426.550 2.65 TRP 17 2.33 3165.578 3.45 TYR 35 4.79 5710.992 6.23 VAL 46 6.29 4560.075 4.98
WARNING:
If the protein doesn't have got hydrogens, the predicted charge isn't shown. If
protein contains special non-aminoacidic groups and/or metal ions, the predicted
charge can be incorrect.
3.8.1.2 Evaluation of interaction energy (molecular docking score)
VEGA can evaluate the ligand-biomacromolecule interaction energy through molecular mechanics calculations. Some scoring functions are implemented (for more details, see -k option).At the present time, only the CVFF force field is implemented. Please remember that ligand and receptor must have correctly assigned charges (see -c option) if you want to calculate the electrostatic interaction. You can specify the dielectric constant with -d option (default 1.0) and the ligand (see -e option). After the energy calculation, VEGA shows (or writes in a XML file) the total interaction energy, the components for each atom and residue.
| Keyword | Description |
| ALCHEMY | Alchemy format. |
| AMMP | AMMP molecular mechanics software. |
BIOSYM |
New Biosym .car file (archive 3). |
| ChemSol | ChemSol 2 solvatation energy software. |
| CIF | IUCr Crystallographic Information Framework. |
| CML | Chemical Markup Language (CML) version 1.0. |
| CML2 | Chemical Markup Language (CML) version 2.0. |
CRD |
CHARMM text file format. |
| CRT |
Indiana University Molecular Structure Center (IUMSC) CRT format for crystallographic structures. |
| CPMDXYZ | CPMD (Car-Parrinello Molecular Dynamics Code) Cartesian output file. |
CSSR |
Cambridge Data File. |
FASTA |
FASTA is not a real molecular file, because it can store only the primary structure of proteins and DNA/RNA sequences. |
| GAMESS | Cartesian GAMESS format. |
| GAUSSIN | Gaussian Cartesian input. |
GROMOS |
This is the special file format of the molecular mechanics package Gromos/Gromacs. |
GROMOSNM |
GROMOS with the coordinates in nanometers. |
IFF |
Interchange File Format. This is a binary file with an AmigaOS chunk structure (like IFF-ILBM, AIFF, etc). All chunks are optional and the structure is totally expandable (see Appendix D). |
| INCHI | IUPAC Chemical Identifier (InChI). |
| INCHIAUX | Same of above with auxiliary data. |
| INFOXML |
This is not a real file molecule file format, because it's a XML container of property data only. The user can select the properties to calculate including the -k[KEYWORDS] option. |
| MDLMOL | MDL Molfile. |
| MMCIF | Crystallographic Information Framework for macromolecules. |
MOL2 |
Tripos Sybyl Mol2 file format. |
| MOPCAR | Mopac cartesian coordinate file (see below). |
MOPINT |
The Mopac internal coordinates file (.dat) is useful to link Mopac with other software packages. The Mopac keyword CHARGE is automatically calculated by atomic charges. Other keywords can be specified with -k[KEYWORDS] option. The preferences file of VEGA (prefs in Data directory) contains a special record Mopac keyword used by default. |
MSF |
MSI Quanta binary file. Its complexity and the poor documentation available have not allowed a full implementation of this format. You can only overwrite an existing MSF file (that must be compatible with the input), but not create a new file. |
| NAMDBIN | NAMD .coor double precision binary coordinate file. |
OLDBIOSYM |
Old Biosym (Accelrys) .car file (archive 1). |
| PDB | PDB pre-2.0 specifications. |
PDB2 |
PDB 2.2 full standard (default). |
| PDBA | PDB full standard with special records to include atomic charges, force field parameters and ATDL description for each atom. It's totally compatible with the PDB standard, because the extra information are placed in REMARK records. |
PDBF |
PDB full standard with special REMARK records to include atomic charges and force field parameters. It's also totally compatible with the PDB standard. |
| PDBL |
The PDB Large file format allows to save molecules with more than 99999 atoms, inserting a TER record after 99999 atoms and restarting the numbering from 1. It's full compatible with the NAMD package and doesn't support the connectivity (CONECT record). |
PDBNOTSTD |
Simplified PDB format, more compatible with software packages that have a partial implementation of Brookhaven specifications. Special records (HETATM, TER, CONECT and MASTER) are not used. |
PDBQ |
PDB full standard with atomic charges placed in the last right column. |
| PDBQT |
AutoDock 4 PDBQT. It's a standard PDB file with two extra columns for charges and potentials. It could contains the information for the torsion angles. |
| PQR |
Modified PDB file with atomic charges and Van der Waals radii in the Occupancy and TempFactor columns. It's the format required by APBS. |
| PQRXML | XML-based format used by APBS. |
| PSFX | PSF topology in X-Plor sub-format required for molecular dynamics (e.g. CHARMM and NAMD). |
| QMC | CSSR variant. |
| RIFF | Interchange File Format (IFF) variant in little endian format (see Appendix D). |
| SMILES | Simplified molecular input line entry specification (SMILES canonical format). |
| SPILLORBS | Spillo Reference Binding Site. |
| VINA | AutoDock Vina PDBQT. It's a standard PDB file with two extra columns for charges and potentials. It could contains the information for the torsion angles. |
XYZ |
Cartesian coordinates file. The first record is the total number of atoms and the next records are for each atom. The atom record contains the element name and X, Y, Z Cartesian coordinates. |
All these output formats are useful for trajectory analysis (see -m [KEYWORDS] option)
| Keyword | Description |
BINPLT |
Generic binary plot. It's a sequence of single precision floats in big endian format. |
CSV |
ASCII text file with each field separated by a semicolon. |
QUANTAPLT |
Accelrys Quanta plot file. |
VEGA can calculate Van Der Waals and accessible to solvent molecular surface. To enable this function, you have to use the -f[OUTPUT_FORMAT] option as shown in the following table:
| Keyword | Type | Description |
| COMFAFLD | Text | COMFA 3D field. When you select this output, you must specify the field type with -m[KEYWORD] option. A Sybyl .rgn file is needed as input also. At the present time, the only implemented filed is vlogP*. |
BIOSYMSRF |
Text |
Van Der Waals and accessible to solvent molecular surface for Insight II package. |
| CSVILM | Text | Molecular hydropathicity index (ILM) surface in CSV (Comma Separated Values) format. |
CSVLOGP* |
Text |
Virtual logP surface in CSV format. |
CSVMEP |
Text |
Molecular Electronic Potential (MEP) in CSV format. |
CSVSRF |
Text |
Van Der Waals and accessible to solvent molecular surface in CSV format. |
| QUANTAILM | Binary | Molecular hydropathicity index (ILM) surface in Quanta format. |
QUANTALOGP |
Binary |
Virtual logP surface in Quanta format. |
QUANTAMEP |
Binary |
Molecular Electronic Potential (MEP) in Quanta format. |
QUANTASRF |
Binary |
Van Der Waals and accessible to solvent molecular surface for Quanta package. |
The default calculation is the water accessible surface (1.4 Å sphere radius). To change the solvent radius (probe), you can use the -g[RADIUS] option. If you set the probe radius to null, VEGA calculates the Van Der Waals surface. The standard point density is 10 for one Å2. See -s[POINTS] option to change this value. Click here if you want more information about the surface calculation method.
In order to support the Web publishing, the Virtual Reality Modeling Language (VRML) was implemented in VEGA. To use this function you can use the -f[OUTPUT_FORMAT] option with the following keywords:
| Keyword | VRML output |
VRML |
VRML 1.0 wireframe representation with standard coloring method. |
VRMLCPK |
VRML 1.0 CPK representation with standard coloring method. |
VRMLPTS |
VRML 1.0 dotted surface representation. |
VRMLSOL |
VRML 1.0 Van Der Waals and accessible to solvent molecular solid surface |
The VRML surface formats can also accept the same options of standard surface outputs (see section 3.7.4).
VEGA can convert the trajectory files of molecular dynamics simulations to different formats. To enable this function, you have to use the -f[OUTPUT_FORMAT] option as shown in the following table:
| Keyword | Type | Compression | Description |
| TRJDCD | Binary | No | CHARMM/NAMD DCD binary file. |
| TRJIFF | Binary | No | IFF/RIFF 64 bit binary file. |
| TRJMOL2 | Text | No | Mol2 multi model. |
| TRJPDB | Text | No | PDB multi model. |
| TRJTRR | Binary | No | Gromacs TRR. |
| TRJXTC | Binary | Yes | Gromacs XTC (lossy compression). |
If you want calculate a surface map with a probe radius different than the default one (the default value is the 1.4Å water radius) without change the prefs file, you can use this option. Please remember that in orded to calculate the Van Der Waals surface, you must set this parameter to zero.
VEGA can solvate a molecule virtually with any type of solvent (e.g. H2O, CCl4,
etc). The cluster file must be placed in Data/Clusters (Data\Clusters) directory
and can be in any VEGA supported format (also packed). This is a solvent assembly with
cubic shape (usually with dimension of 50x50x50 Å ), optimized, with uppercase file name
without extension (e.g. WATER, CCL4, etc).
SHELL is the solvent cluster name (e.g. WATER). SHAPE is the form of solvatation cluster:
BOX for cubic clusters, SPHERE for spherical clusters and LAYER to solvate with a layer of
solvent. RAD is a value in Å that followed by BOX, defines the box side, by SPHERE, the
sphere radius and by LAYER the layer thickness.
This option define the torsion angles in the molecule. It can be used with the file formats that require the torsions (e.g. AutoDock's PDBQT).
| Argument | Description |
| ALL | Define all possible torsions. |
| AUTODOCK | Define the flexible torsions for AutoDock 4. |
| FLEX | Define the flexible torsions only. |
This option is useful to pass the control keywords when the Info XML (-f NFOXML option) or the Mopac (-f MOPINT option) or the Score (-f Score option) format is selected. Remember to use quotas (") if the number of keyword is more than one. In the prefs file, you can specify the default Mopac keywords. The Info XML keywords are summarized in the following table:
| Keyword | Calculated property |
| AACOMP | Amino acid composition (occurrence, occurrence percentage, mass, mass percentage, protein mass, protein mass percentage, number of amino acids). |
| ALL | All properties (default option). |
| ANGLES | Number of bond angles. |
| AREA | Surface area and surface diameter. |
| ATOMS | Number of atoms. |
| ATMTYPES | Atom types and occurrences of atom types. |
| BONDS | Number of bonds. |
| CENTGEO | Geometric center. |
| CENTMASS | Center of mass. |
| CENTROIDS | Number of centroids. |
| CHAINS | Number of chains |
| CHARGE | Total charge. |
| CHIRALATMS | List of the chiral atoms. |
| CHIRALNUM | Number of the chiral atoms. |
| DIMENSIONS | Molecule dimensions. |
| DIPOLE | Dipole moment. |
| EZBONDS | List of the bonds with E/Z geometry. |
| EZNUM | Number of the bonds with E/Z geometry. |
| FORMULA | Molecular formula. |
| GCMR | Molar refractivity (Ghose & Crippen method). |
| GYRRAD | Radius of gyration. |
| HBONDACC | Number of H-bond acceptors (N and O only). |
| HBONDDON | Number of H-bond donors (H-N and H-O only). |
| HEAVYATOMS | Number of heavy atoms. |
| HLB | Davies, Griffin, PSA-based and mean hydrophilic-lipophilic balances (HLBs). |
| HYDROGENS | Number of hydrogens. |
| ISOTOPIC | Isotopic distribution (isotopic pattern). Format: mass probability (%) |
| LOGPCRIPPEN | Ghoose & Crippen logP and lipole. |
| LOGPBROTO | Broto & Moreau logP and lipole. |
| LOGPVIRTUAL | Bernard Testa's virtual logP. |
| MIMASS | Monoisotopic mass. |
| MOLECULES | Number of molecules. |
| MOLNAME | Molecule name. |
| PROBERAD | Probe radius used in the surface calculation (AREA). |
| PSA | Polar and apolar surface areas. |
| RESIDUES | Number of residues. |
| SEGMENTS | Number of segments. |
| SMILES | SMILES string. |
| TORADOCKNUM |
Number of flexible torsions used by AutoDock to perform the in situ conformational search. |
| TORFLEXNUM | Number of flexible torsions. |
| TORNUM | Number of torsions. |
| VOLUME | Molecular volume and volume diameter. |
| WATERS | Number of waters. |
| WEIGHT | Molecular weight. |
All these keywords can be combined separating them by a space character.
The Score keywords that can be used to select one or more score functions, are summarized in the following table:
| Keyword | Score function |
| CHARMM | R6-R12 non-bond interaction evaluated by CHARMM 22 force field provided by Accelrys. To perform this calculation, the parm.prm file must be copied in the ...\VEGA\Data\Parameters directory. This file is not included in the package for copyright reasons. |
| CHARMM22 | R6-R12 non-bond interaction evaluated by CHARMM 22 force field. |
| CHARMM36 | R6-R12 non-bond interaction evaluated by CHARMM 36 force field. |
| CONTACTS |
The scores are evaluated by counting the number of ligand/receptor
contacts and by normalizing it by the number of heavy atoms and the mass
of the ligand. Moreover, if the receptor is a protein, it generates an
interaction fingerprint with a size of 20 bits (one bit for each amino
acid type) and a contact map in which the number of contacts per amino
acid type is reported. To determine if there is a contact between a pair
of atoms, the distance between the two centres is calculated and if it
is less than 2.5 Å, then there is a contact. This
threshold value can be changed in the prefs
file. If -o option is used, the resulting XML file
will include the also the two additional scores with extra tag
(attributes: id = 1
|
| CVFF | R6-R12 non-bond interaction evaluated by CVFF force field. |
| ELECT | Electrostatic interaction. To change the dielectric constant value, use the -d option. |
| ELECTDD | Distance-dependent electrostatic interaction. To change the dielectric constant value, use the -d option. |
| MLPINS | Hydrophobic interaction calculated using the Broto's and Moreau's atomic constants*. |
| MLPINS2 | Hydrophobic interaction in which the distance between interacting atom pairs is considered as square value*. |
| MLPINS3 | Hydrophobic interaction in which the distance between interacting atom pairs is considered as cube value*. |
| MLPINSF | Hydrophobic interaction in which the distance is evaluated by the Fermi's equation*. |
All these keywords can be combined separating them by a space character also.
* From Vitoli G. et al., Bioorg. Med. Chem. 18 (2010) 320-19.
"The MLP Interaction Score (MLPInS) is computed using the atomic fragmental system proposed by Broto and Moreau and a distance function that define how the score decrease with increasing distance between interacting atoms. In detail, the equation to compute such an interaction score is reported below:
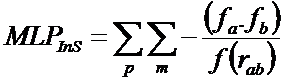
where fa and fb denote the lipophilicity increments for a pair of atoms and rab is the distance between them. The first sum (p) concerns all ligand’s atoms and the second (m) all enzyme’s atoms. The basic assumption in the calculation of the MLPInS, which encodes the contributions of the various intermolecular forces measured experimentally in partition coefficients, is that the score is favourable (i.e. negative) when both increments have the same sign (as denoted by the negative sign in in the equation), or unfavorable (repulsive forces) when the score has a positive sign. When the atomic parameters are both positive, MLPInS encodes hydrophobic interactions and dispersion forces, the importance of which is well recognized in docking simulations, and it accounts for polar interactions, in particular H-bonds and electrostatic forces when the atom ic parameters are both negative".
This command adds the hydrogens to the loaded molecule/s, saturating all atom valences. MOLTYPE is the molecule type and it can be:
| MolType | Description |
| GEN | Generic organic molecule. |
| GENBO | Generic organic molecule, bond order algorithm. |
| NA | Nucleic acid. |
| NABO | Nucleic acid, bond order algorithm. |
| PROT | Protein. |
| PROTBO | Protein, bond order algorithm. |
Use the bond order algorithm if the molecule geometry is uncertain (e.g. raw 3D structure or 2D structure), but it works well only if the bond order is correctly assigned.
This option allows to do measures for each frame or to extract one or more frames of a molecular dynamics trajectory file. You must specify a keyword to set the kind of measure and optionally the atom selection:
| Keyword | Description |
| ANGLE A1 A2 A3 | Bond angle. |
| DISTANCE A1 A2 | Bond length. |
| DIPOLE | Molecular dipolar moment. |
| EXTRACT F1 [F2] |
Extract one ore more molecules from the trajectory file starting from the F1 frame to the F2 frame. F2 is optional and if it's omitted, the extraction proceed until the last frame. |
| GYRRAD | Gyration radius. |
| ILM | Molecular hydropathicity index (water cluster required). |
| LIPOLEBR | Lipole (Broto & Moreau) |
| LIPOLECR | Lipole (Ghoose & Crippen) |
| SURFACE A1 ... | Surface area. |
| SURFDIA A1 ... | Surface diameter. It's the diameter of a theoretical sphere with the surface area of the molecule. |
| OVALITY | Ovality. It's calculated by the following equation:
where: O = ovality; A = area; V = volume |
| PLANEANG A1 A2 A3 A4 A5 A6 | Angle between planes defined by A1, A2, A3 and A4, A5, A6. |
| PSA | Polar surface area. |
| RMSD | Calculates the RMSD between the first trajectory frame and the others excluding the hydrogens. |
| RMSDH | As above but including the hydrogens. |
| RMSDALN | Aligns the the first trajectory frame with the others and calculates the RMSD excluding the hydrogens. |
| RMSDALNH |
As above but including the hydrogens. This keyword is equivalent to the old RMSD until the 3.2.2 version. |
| RMSDSYMCOR |
It performs the RMSD calculation without any alignment, but considering the symmetric atoms as equivalent. To do the atom pair selection, the Cahn-Ingold-Prelog (CIP) weights are assigned to each atom and than the hungarian algorithm (also known as Munkres algorithm or Kuhn-Munkres algorithm) is applied to to compute the optimal assignment, minimizing the total cost. |
| RMSDSYMCORH | As above but including the hydrogens. |
| TORSION A1 A2 A3 A4 | Torsion angle. |
| VLOGP | Virtual logP. |
| VOLUME | Molecular volume. |
| VOLDIA | Volume diameter. It's the diameter of a theoretical sphere with the volume of the molecule. |
To select each atom required in the mesure (e.g. A1 A2 etc), you must use the atom number only, or the following syntax: ATOM:RESNAME:RESNUM. RESNAME and RESNUM are optional if ATOM is univocal. Suppose to have a benzene ring and you would like indicate the third atom, like shown in the following PDB file:
...
ATOM 2 C2 BEN 1
-0.695 1.203 -0.002 1.00 0.00
ATOM 3 C3 BEN
1 -1.389 0.000
-0.006 1.00 0.00
ATOM 4 C4 BEN 1
-0.695 -1.203 -0.007 1.00 0.00
...
you can use, without differences, 3 or C3 or C3:BEN or C3:BEN:1. If you want select the atom 482 in a polypeptidic sequence where only one proline is present, you can indicate it with 482 or CA:PRO or CA:PRO:32, but not CA only:
... ATOM 481 N PRO 32 -29.658 -2.153 7.524 1.00 0.00 ATOM 482 CA PRO 32 -28.294 -1.798 7.139 1.00 0.00 ATOM 483 C PRO 32 -27.169 -2.471 7.908 1.00 0.00 ... ATOM 495 N VAL 33 -25.978 -2.393 7.325 1.00 0.00 ATOM 496 CA VAL 33 -24.749 -2.884 7.927 1.00 0.00 ATOM 497 C VAL 33 -23.841 -1.699 7.661 1.00 0.00 ...
If more than one proline is present in this sequence, you can't use CA:PRO neither.
At the end of the property calculation, VEGA shows the ranges, the average value and the standard deviation. If you want exclude the influence of the water in the calculation of dipolar moment, molecular surface, Virtual logP and molecular volume, you can use the -w option.
This switch enables the normalization of atomic coordinates. The geometry center of a single molecule or a complex is moved to the origin of Cartesian axes.
With -o parameter, you can specify the name of the output file with or without extension. If the filename doesn't have any extension, VEGA automatically adds the appropriate one on the basis of the selected output format (see -f option). The most common extension used by VEGA are shown in the following table:
| Extension | Type | Add | File format |
| .alc | T | Y | Alchemy. |
| .amp | T | Y | AMMP. |
.arc |
T |
N |
Mopac optimized internal coordinates. |
.car |
T |
Y |
Accelrys CAR file (old and new subformat). |
| .cif | T | Y | IUCr Crystallographic Information Framework (CIF/mmCIF). |
| .cml |
T |
Y |
Chemical Markup Language (CML). |
.cor |
T |
Y |
Accelrys CAR file with optimized coordinates. |
.crd |
T |
Y |
CHARMM. |
| .crt | T | A | IUMSC CRT. |
| .cs | T | Y | ChemSol 2. |
.cssr |
T |
Y |
Cambridge Data File (CSSR). |
| .csv | T | Y | Surface in CSV format. |
.dat |
T |
Y |
Mopac cartesian/internal coordinates. |
.dcd |
B |
Y |
CHARMM/NAMD trajectory file. |
.ene |
T |
N |
Accelrys CHARMm energy file. |
.ene |
T |
Y |
VEGA interaction energy file. |
.ent |
T |
N |
PDB. |
.fas |
T |
Y |
FASTA. |
| .fld | T | Y | Tripos COMFA field. |
.gro |
T |
Y |
Gromos/Gromacs. |
.iff |
B |
Y |
Interchange File Format (IFF). |
| .inc | T | N | InChI. |
| .inchi | T | Y | InChI. |
.inf |
T |
Y |
VEGA information file. |
| .inp | T | Y | GAMESS cartesian. |
| .log | T | Y | Gaussian output. |
.ml2 |
T |
Y |
Tripos Sybyl Mol 2. |
| .mol | T | Y | MDL Molfile (V2000), MDL Extended Molfile (V3000). |
.msf |
B |
Y |
MSI Quanta. |
.par |
T |
N |
VEGA parameters. |
.pdb |
T |
Y |
PDB, PDB2, PDBA, PDBF, PDBL and PDBQ. |
| .pdbqt | T | Y | AutoDock 4 / Vina PDBQT. |
| .pqr |
T |
T |
PQR. |
| .psf | T | Y | PSF and PSF X-Plor. |
.qmc |
T |
N |
QMC (CSSR like format). |
| .smi | T |
Y |
Smiles. |
.srf |
B |
Y |
Accelrys Quanta surface. |
.srf |
T |
Y |
Accelrys Insight surface. |
.tem |
T |
N |
VEGA template. |
.wrl |
T |
Y |
VRML (Virtual Reality Markup Language). |
| .xml |
T |
Y |
PQR XML. |
| .xyz | T | Y | CPMD XYZ. |
| .xyz | T | Y | TINKER XYZ. |
.xyz |
T |
Y |
XYZ. |
Where the column Extension is the file extension, Type is
the file type (T = text, B = binary), Add shows if VEGA adds automatically the
extension and File Format is the name of file format.
If you execute VEGA without -o parameter, the output is redirected to the
console (stdout) or to a special device driver (e.g. PRT: for AmigaDOS). This function is
very useful to interface VEGA with another program that can get the input from console.
The redirection is possible with text file formats only.
This function allows to assign the atom types using a specified force field template. This is the most complex function implemented in VEGA. The first challenge being the creation of an universal language, called ATDL (Atom Type Description Language) able to describe virtually any atom type. For more information about ATDL, click here. VEGA uses the force field template files stored in Data directory with the extension .tem (lowercase). The name of these files must be uppercase, but the argument of -p option is case-insensitive. In order to assign the correct atom types, VEGA uses a multiple step algorithm:
Although these steps are very complex, the total process speed is very high.
Fix the bond order using the specified method that could be: ALL (find the order of all bond) or RINGS (fix the bonds of the aromatic rings making them partial double).
This switch removes the hydrogen atoms: the empty or ALL arguments remove all hydrogens and the APOLAR removes the apolar hydrogens only.
With this parameter you can change the point density of a surface map. POINTS is the number of points per surface unit (Å2). The default value is stored in the prefs file and usually it is set to 10. For more information about surface calculation, please see the -f[FORMAT] option.
The -t option allows to change the protein secondary structure. Two operational mode are available: in the former the user assigns Phi, Psi and Omega torsion values by the syntax TORSION_NAME=value (e.g. Phi=-135), in the latter he put secondary structure name as reported in the following table:
| Sec. structure name | Code | Phi | Psi | Omega | Description |
| AlphaHelix | H | -57.8° | -47.0° | 180.0° | Alpha helix (3,6.13). |
| LeftHelix | L | 57.8° | 47.0° | 180.0° | Left handed alpha helix. |
| 310Helix | 3 | -74.0° | -4.0° | 180.0° | 3.10 helix. |
| PiHelix | P | -57.1° | -69.7° | 180.0° | Pi helix |
| Beta | E | -135.0° | 135.0° | 180.0° | Generic beta strand. |
| BetaAnti | A | -140.0° | 135.0° | 180.0° | Beta strand in anti-parallel sheet. |
| BetaPar | B | -120.0° | 115.0° | 180.0° | Beta strand in parallel sheet. |
Through the keyword PATTERN=, you can set the secondary structure for each residue according the previous table (Code column). If you specify U code, Phi, Psi and Omega are retrieved from the user-defined values set as explained above.
This option can be used to assign the secondary structure when a Fasta file is loading and if it's omitted, the generic beta strand structure is assigned. All sub-parameters are case insensitive.
This command adds the side chains to a protein. The side chain database is placed in the Data/Fragments directory and it's called Amino acids L.zip. The side chains are added without hydrogens and so, if you need them, you must use the -l option also.
Set the number of CPUs used in the parallel calculations. The 0 argument means that all installed CPUs are used.
This switch removes all the water molecules present in an assembly. Please note that VEGA
do not find the water molecules by residue names (e.g. HOH, TIP3, etc), but on the basis
of connectivity table. This approach is slower but more precise and independent of residue
naming.
You can use the -w option in trajectory analysis to neglect the water influence in the
evaluation of dipolar moment, molecular surface and Virtual logP.
It extracts a molecule from the input database that must be in SDF or ZIP format. The arguments of this options can be:
| Argument 1 | Argument 2 | Description |
| LIST | - | List the name of the molecules in the database. |
| NAME | molecule name | Extract the molecule with the specified name. |
| NUM | molecule number | Extract the molecule with the specified identification number. |
Find the molecules in the assembly using the connectivity information. This feature is useful when you need to select the molecule (ligand) in the interaction energy evaluation (see -e and -k options), because all file formats, excluding IFF/RIFF, can't store molecule information (starting and ending atoms) in the atom list.
Add the capping to N- and C-term position of a peptide when it is built from its primary sequence while is loaded from a FASTA file.