To calculate one or more docking scores, you must select the ligand, typing its molecule number in the Ligand field or clicking on the ligand in the main window.
8.9 Evaluation of the interactions (docking score analysis)
In order to analyze the docking results, you can
calculate the interaction energy between a ligand and a biomacromolecule, evaluating the contributes of
each residue that can be shown in the graphic environment. This function is available
by choosing Calculate
![]() Interactions
from the main menu.
Interactions
from the main menu.
To calculate one or more docking scores, you must select the ligand, typing its
molecule number in the Ligand field or clicking on the ligand in the
main window.
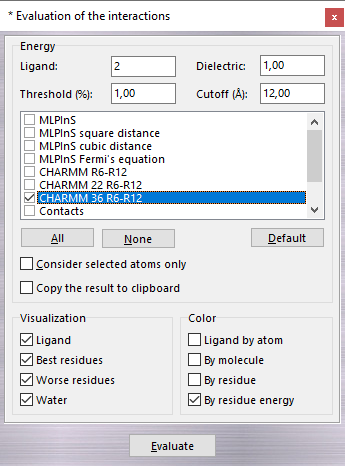
The Dielectric (dielectric constant) parameter is used in the electrostatic energy evaluation only (Electrostatic and Electrostatic distance dependent) and the Cutoff field defines the maximum distance between an atom pair over which the contribution is discarded. You can select more than one or scoring functions as shown in the following table:
| Scoring function | Description |
| CHARMM R6-R12 | R6-R12 non-bond interaction evaluated by CHARMM 22 force field provided by Accelrys. To perform this calculation, the parm.prm file must be copied in the ...\VEGA ZZ\Data\Parameters directory. This file is not included in the package for copyright reasons. |
| CHARMM 22 R6-R12 | R6-R12 non-bond interaction evaluated by CHARMM 22 force field. |
| CHARMM 36 R6-R12 | R6-R12 non-bond interaction evaluated by CHARMM 36 force field. |
| CONTACTS | The scores are evaluated by counting the number of ligand/receptor contacts and by normalizing it by the number of heavy atoms and the mass of the ligand. Moreover, if the receptor is a protein, it generates an interaction fingerprint with a size of 20 bits (one bit for each amino acid type) and a contact map in which the number of contacts per amino acid type is reported. To determine if there is a contact between a pair of atoms, the distance between the two centres is calculated and if it is less than 2.5 Å, then there is a contact. This threshold value can be changed in the Preferences window. |
| CVFF R6-R12 | R6-R12 non-bond interaction evaluated by CVFF force field. |
| Electrostatic | Electrostatic interaction. |
| Electrostatic distance dependent | Distance-dependent electrostatic interaction. |
| MLPInS | Hydrophobic interaction calculated using the Broto's and Moreau's atomic constants. |
| MLPInS square distance | Hydrophobic interaction in which the distance between interacting atom pairs is considered as square value. |
| MLPInS cubic distance | Hydrophobic interaction in which the distance between interacting atom pairs is considered as cube value. |
| MLPInS Fermi's equation | Hydrophobic interaction in which the distance is evaluated by the Fermi's equation. |
For more information about MLPInS, click here.
Checking Best residues, the residues with a partial
interaction energy greater than Threshold (%) of the total energy are
shown in graphic
window. If more than one scoring function is selected, it will be
impossible to show the best interacting residues in the main window. Checking Worse residues,
the residues with the worse energy contribution are shown. Checking Consider
selected atoms only, the active (visible) atoms are considered only in the
calculation and checking Copy the results to clipboard, the evaluated
energies are copied to clipboard. This feature is useful to paste the results to
a spreadsheet (e.g. Microsoft Excel) to elaborate them.
Your can modify the visualization choosing the
colouring method (Ligand by atom, By molecule, By residue and
By residue energy) and
which residues to show (Ligand, Best residues, Worse
residues and Water
molecules).
An output example is shown below:
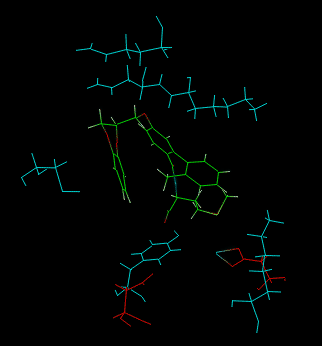 |
Cyan = best residues Red = worst residues Green = ligand |
WARNING:
To evaluate the interaction energy, you must follow these rules: